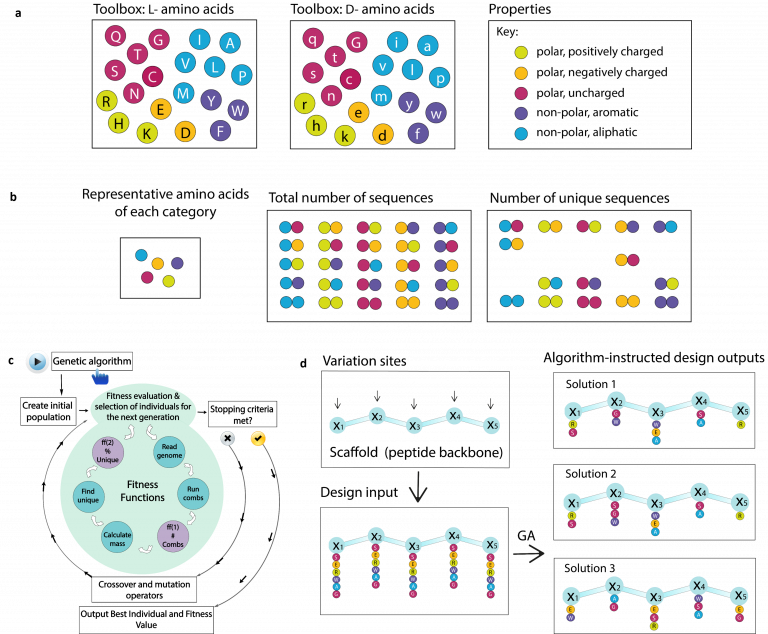
Random peptide libraries that cover large search spaces are often used for the discovery of new binders, even when the target is unknown. To ensure an accurate population representation, there is a tendency to use large libraries. However, parameters such as the synthesis scale, the number of library members, the sequence deconvolution and peptide structure elucidation, are challenging when increasing the library size. To tackle these challenges, we propose an algorithm-supported approach to peptide library design based on molecular mass and amino acid diversity. The aim is to simplify the tedious permutation identification in complex mixtures, when mass spectrometry is used, by avoiding mass redundancy. For this purpose, we applied multi (two- and three-)-objective genetic algorithms to discriminate between library members based on defined parameters. The optimizations led to diverse random libraries by maximizing the number of amino acid permutations and minimizing the mass and/or sequence overlapping. The algorithm-suggested designs offer to the user a choice of appropriate compromise solutions depending on the experimental needs. This implies that diversity rather than library size is the key element when designing peptide libraries for the discovery of potential novel biologically active peptides.
